The CQLS is pleased to announce our new Illumina MiSeq i100 sequencer. As Illumina phases out their original MiSeq we have upgraded to the new MiSeq i100. The CQLS MiSeq i100 sequencing service is now available.
Illumina’s new MiSeq is a lower output sequencer compared to the NextSeq or NovaSeq. The new MiSeq i100 offers similar flow cell sizes to the original MiSeq with greatly reduced sequencing costs and additional sequencing lengths, including 500bp paired end runs. Updated prices are found on our fee site: CQLS Service Fees.
Illumina’s MiSeq i100 is a patterned flow cell like the NextSeq. Patterned flow cells are subject to low levels of index hopping. We suggest reviewing and adopting the mitigation techniques outlined by Illumina: “Minimize index hopping in multiplexed runs.” Please contact us with questions.
CQLS will host an information session with Illumina on Friday, February 20th at 12:30. Food will be provided. Please join us to learn about the MiSeq i100.
The CQLS’ original MiSeq service contract will end on June 30th. Please contact us if you would like help transitioning experiments to the new MiSeq i100.
The Illumina MiSeq i100 purchase was made possible by the DRI 2025 Research Equipment Reserve Fund.
Over the past year CQLS moved HPC users to the new Wildwood HPC. This new HPC included significant upgrades to management servers, operating systems, queuing software (SLURM), ONID authentication, a larger all-access queue (all.q), and custom job submission software (hpcman). Since this transition the CQLS has been working with the other HPC groups on campus to create a consolidated campus-wide HPC and provide unified HPC experience to users, and accessible research computing across OSU.
The College of Earth, Ocean and Atmospheric Sciences (CEOAS) has completed consolidation of users and HPC resources into the Wildwood HPC. The CQLS and CEOAS HPC clusters now share login and management servers, storage, and all.q. The CQLS is currently working with Advanced Research Computing Services (ARCS) and the College of Engineering (COE) to join their HPC clusters with the Wildwood HPC.
The Wildwood HPC has grown in computing capacity and storage. There are approximately 8000 CPUs and 65 GPUs available on the all.q and in priority queues. The all-access queue, all.q, has 480 CPUs and also has an available GPU queue and continues to grow as we intake more users. The Wildwood HPC has 12.3PB of network-attached storage.
The CQLS has also rebuilt the Advanced Cyberinfrastructure Teaching Facility (ACTF), an HPC teaching cluster. The ACTF HPC mimics the Wildwood HPC but has special class-specific resources and areas for students and instructors to work together. Instructors are able to teach linux-based programming and analysis classes and access student directories to check assignments. Please contact us if you are interested in utilizing this resource.
HPC upgrades and changes have necessitated downtimes of servers and storage and we appreciate user’s patience and flexibility for as we continue to make improvements. OSU is decommissioning the Kerr Administration server room where the Wildwood storage servers reside. We are helping the University to vacate the Kerr server room by moving Wildwood HPC storage to a new location in Burt Hall.
The CQLS was awarded a 2025 Research Equipment Reserve Fund (RERF) to purchase a new Illumina MiSeq i100.
The CQLS has run an Illumina MiSeq for 12 years as the center of a highly successful low-range sequencing service. Primarily used for microbiome, environmental, and amplicon samples, the MiSeq offers a lower volume of Illumina high-throughput sequencing compared to the CQLS’ Illumina NextSeq. Illumina is transitioning the original MiSeq to end-of-life service. By upgrading to the new MiSeq i100, the CQLS will replicate our current sequencing abilities while drastically reducing instrument maintenance costs for the CQLS and consumable costs for OSU researchers.
The Illumina MiSeq i100 is a next-generation sequencer that sequences low-volume flow cells, 5 Million sequences in 300 or 600 base-pair lengths or 25 Million sequences in 100, 300, or 600 base-pair lengths. The MiSeq i100 is quicker and cheaper to run than other short-read sequencers.
Key features include:
Cost efficiency: Cost-effective consumables enable more affordable sequencing
Speed: Dramatic reduction in run times: as fast as four hours, with same-day results (4× faster than MiSeq)
Room-temperature shipping and storage for reagents: Allowing for greater flexibility to sequence on demand without the need to thaw reagents
Simplicity: Simpler, streamlined operations for various levels of sequencing experience
The new MiSeq i100 is upgraded from the original MiSeq in several ways. Flow cells are lower-cost and higher volume so researchers will be able to obtain more sequence at a lower price. The small volume sequencing (1M) of the original MiSeq is no longer offered but researchers will be able to sequence higher volume runs at a lower or similar price. Running the instrument is also more streamlined: run times are 4x faster; many reagents and consumables are now stored in room temperature; the instrument is simpler to maintain resulting in a lower yearly service contract price.
Bing Wang, CQLS; Marco Corrales Ugalde HMSC; Elena Conser HMSC
By leveraging the scalability and parallelization of high-performance computing (HPC), CQLS and CEOAS Research Computing, collaborated with PhD student Elena Conser and postdoctoral research scholar Marco Corrales Ugalde from Hatfield Marine Science Center’s (HMSC) Plankton Ecology Laboratory to complete the end-to-end processing of 128 TB of plankton image data collected during Winter and Summer of 2022 and 2023 along the Pacific Northwest coast of the United States (Figure 1a) .The workflow, which encompassed image segmentation, training library development, and custom convolutional neural network (CNN) model training, produced taxonomic classification and size data for 11.2 billion plankton and particle images. Processing was performed on the CQLS and CEOAS HPC and Pittsburgh Supercomputing Center Bridges-2, consuming over 40,000 GPU hours and 3 million CPU hours. CQLS and CEOAS Research Computing provided essential high-performance computing resources, such as NVIDIA Hopper GPUs, IBM PowerPC system, and multi-terabyte NFS storage. In addition, these computing centers contributed with software development that expedited the processing of images through our pipeline, such as custom web applications for image visualization and annotation, and automated, high-throughput pipelines for segmentation, classification, and model training.
Project introduction
Plankton (meaning “wanderer,” in Greek) are composed of a diverse community of single-celled and multicellular organisms, comprising an extensive size range from less than 0.1 micrometers to several meters. Planktonic organisms also vary extensively in their taxonomy, from bacteria and protists to the early life stages of fishes and crustaceans. The distribution and composition of this diverse “drifting” community is determined in part by the ocean’s currents and chemistry. Marine plankton form the foundation of oceanic food webs and play a vital role in sustaining key ecosystem services, such as climate regulation via the fixation of atmospheric CO2, the deposition of organic matter to the ocean’s floor, and by the production of biomass at lower levels of the food chain, which sustains global fisheries productivity. Thus, understanding plankton communities is critical for forecasting the impacts of climate change on marine ecosystems and evaluating how these changes may affect climate and global food security.
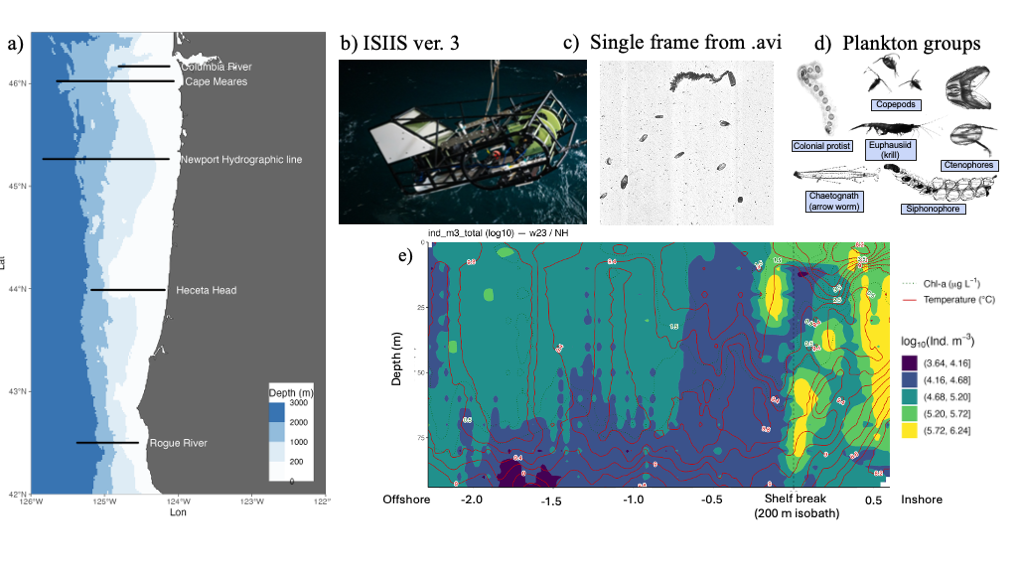
Figure 1. Plankton imagery was collected along several latitudinal transects through the Pacific Northwest coast (a), by the In-situ Ichthyoplankton Imaging System (ISIIS) (b). This data is split into frames (c) and then individual plankton and particle images (d). The high image capture rate (20 frames/second) along a large spatial range allows to provide a detailed description of plankton abundance patterns (colors in panel e) and their relation to oceanographic features (Chlorophyll concentration, dotted isolines, and Temperature, red isolines).
HMCS’s Plankton Ecology Lab, in collaboration with Bellamare engineering, has been the lead developer of the In-situ Ichthyoplankton Imaging System (ISIIS), a state-of-the-art underwater imaging system (Figure 1b) that captures in situ, real-time images of marine plankton (Figures 1c-d). It utilizes a high-resolution line-scanning camera and shadowgraph imaging technology, enabling imaging of up to 162 liters of water per second with a pixel resolution of 55 µm, and detecting particles ranging from 1 mm to 13 cm in size. In addition, ISIIS is outfitted with oceanographic sensors that measure depth, oxygen, salinity, temperature, and other water characteristics. ISIIS is towed behind a research vessel during field deployments. In a single day, a vessel may cover 100 miles while towing ISIIS. ISIIS collects imagery data at a rate of about 10 GB per minute with its paired camera system. A typical two-week deployment produces datasets containing more than one billion particles and approximately 160 hours of imagery, resulting in over 35 TB of raw data. This unprecedented volume of data, together with its high spatial resolution and simultaneous sampling of the environmental context (Figure 1e) allows to address questions of how mesoscale and submesoscale oceanography determine plankton distribution and food web interactions.
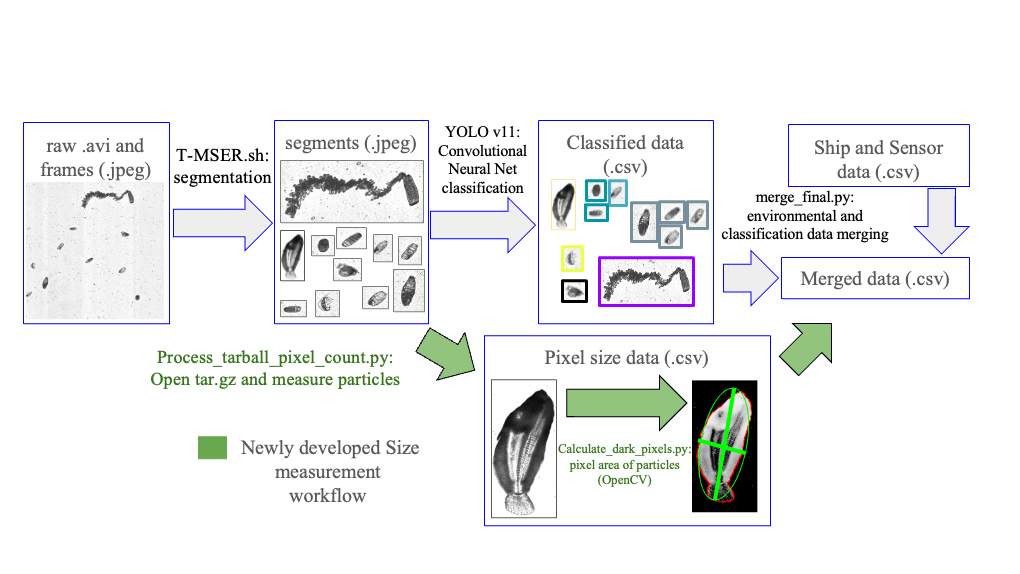
Processing this vast amount of data is complex and highly demanding on computing resources. To address this challenge, CQLS and CEOAS Research Computing developed a comprehensive workflow encompassing image segmentation, training library construction, CNN model development, and large-scale image classification and particle size calculation. In the first step, raw imagery is segmented into smaller regions containing regions of interest. These regions are subsequently classified using a CNN based model (Figure 2). In earlier work, a custom sparse CNN model was developed for classification, but thanks to significant advances in computer vision, more efficient models such as YOLO (“You Only Look Once”) have been developed. To train a new YOLO plankton model, a training library of 72,000 images across 185 classes was constructed. Additionally, a test library of 360,000 images was curated to verify model accuracy and ensure robust performance. This improved workflow was applied to 128 TB of ISIIS video data, enabling the classification and size measurement of 11.2 billion images.
Figure 2. Data processing workflow from raw video to training library development, model training, and plankton classification and particle size estimation
Hardware and software support from CQLS and CEOAS research computing
CQLS and CEOAS Research Computing provide high-performance computing resources for research and is accessible to all OSU research programs. The HPC has a capacity of 9PB for data storage, 45TB of memory, 6500 processing CPUs, and 80+ GPUs. The CQLS and CEOAS HPC can handle more than 20,000 submitted jobs per day.
1. GPU resources
Multiple generations of NVIDIA GPUs are available across architectures on the CQLS and CEOAS HPC, including Tesla V100, Grace Hopper, GTX 1080, and A100, with memory capacities ranging from 11 GB to 480 GB as listed below. GPUs are indispensable for image processing and artificial intelligence, delivering orders of magnitude faster performance than CPUs.
In this project, GPU resources were employed to train YOLO models and to perform classification and particle size estimation of billions of plankton images.
cqls-gpu1 (5 Tesla V100 32GB GPUs), x86 platform
cqls-gpu3 (4 Tesla T4 16GB GPUs), x86 platform
cqls-p9-1 (2 Tesla V100 16GB GPUs), PowerPC platform
cqls-p9-2 (4 Tesla V100 32GB GPUs), PowerPC platform
cqls-p9-3 (4 Tesla V100 16GB GPUs), PowerPC platform
cqls-p9-4 (4 Tesla V100 16GB GPUs), PowerPC platform
ayaya01 (8 GeForce GTX 1080 Ti 11GB GPUs), x86 platform
ayaya02 (8 GeForce GTX 1080 Ti 11GB GPUs), x86 platform
coe-gh01, grace NVIDIA GH200s, 480 GB memory, ARM platform
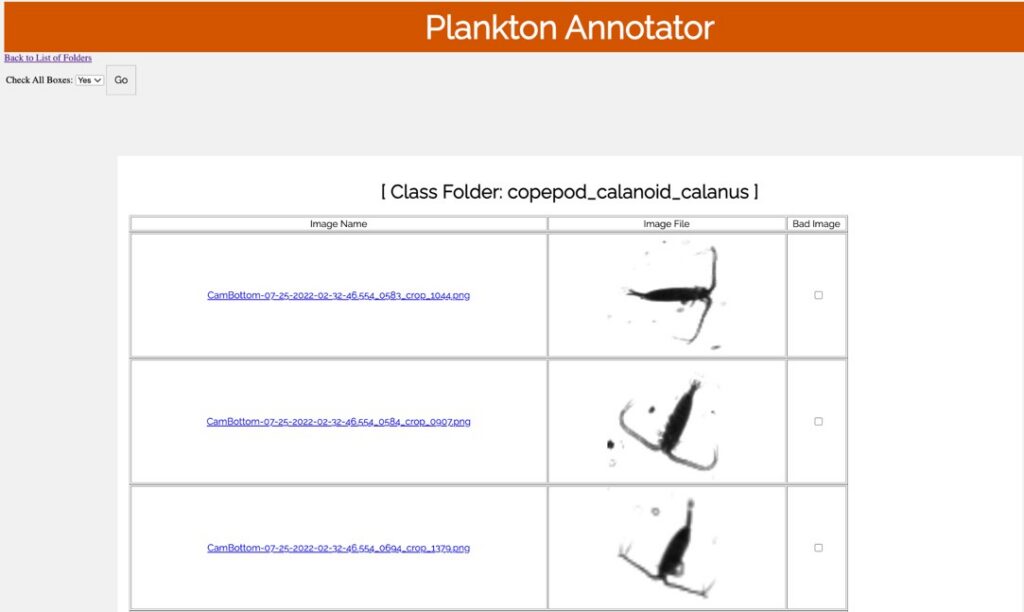
Plankton Annotator (Figure 3) is a web application developed by Christopher M. Sullivan, Director of CEOAS Research Computing. The tool provides an intuitive platform for the visualization and annotation of plankton imagery. The CQLS and CEOAS Research Computing has the capacity for developing and hosting various web applications. In this project, the Plankton annotator was used to efficiently visualize and annotate plankton images, supporting the creation of reliable training library for downstream YOLO models training.
Figure 3. Plankton annotator web application
4. Pipeline developments
CQLS and CEOAS Research Computing has a team of consultants with various programming expertise such as Shell, Python, R, JavaScript, SQL, PHP, and C++. They are available to collaborate on grant proposals, project discussions, experimental design, pipeline development, and data analysis for a wide range of projects in bioinformatics and data science. For this project a series of automated, high-throughput pipelines were updated and created to support image segmentation, model training, species classification and particle size estimation. These pipelines were designed to handle large volumes of data efficiently, ensuring scalability, and reproducibility.
5. Storage
In this project, CQLS and CEOAS HPC provided more than 200 TB of centralized NFS storage to accommodate both raw and processed data. This shared storage infrastructure ensures reliable data access, supports efficient file sharing across the HPC environment, and enables seamless collaboration among researchers.
References
Conser, E. & Corrales Ugalde, M., 2025. The use of high-performance computing in the processing and management of large plankton imaging datasets. [Seminar presentation] Plankton Ecology Laboratory, Hatfield Marine Science Center, Oregon State University, Newport, OR, 19 February.
Schmid, M.S., Daprano, D., Jacobson, K.M., Sullivan, C., Briseño-Avena, C., Luo, J.Y. & Cowen, R.K., 2021. A convolutional neural network based high-throughput image classification pipeline: code and documentation to process plankton underwater imagery using local HPC infrastructure and NSF’s XSEDE. National Aeronautics and Space Administration, Belmont Forum, Extreme Science and Engineering Discovery Environment, vol. 10.
Oregon State University, 2025. Oregon State University Research HPC Documentation. Viewed 18 September 2025, https://docs.hpc.oregonstate.edu.
Sherlock Secure Cloud for Sensitive Research Data Now Available
Researchers conducting research using and analyzing health data often require access to sensitive data such as personally identifiable information (PII), protected health information (PHI), and other controlled unclassified information (CUI). Now after working collaboratively with the Office of Information Security (OIS), University Information Technology (UIT) and the Advanced Research Computing Services (ARCS), OSU researchers have access to a secure cloud environment, known as Sherlock Secure Cloud, for use on a fee basis.
Sherlock Secure Cloud is versatile and customizable for project needs. Customization that covers data collection to analysis and collaboration with others, is possible, all on a fee basis. For example, research that requires collection of sensitive data, using REDCap (Research Electronic Data Capture), is possible, as well a variety of statistical analyses tools can be installed. Project spaces are unique to the approved research team members.
Sherlock Cloud provides a managed multi-Cloud solution that complies with Federal Information Security Management Act (FISMA), Health Insurance Portability and Accountability Act (HIPAA) and NIST CUI requirements. Sherlock Cloud recently achieved a successful SOC2 Type2 certification and follows the NIST 800-53 (Mod) framework that spans over 200 technical, security and administrative controls. These security features are in line with those required for the most common research data use agreements.
CQLS staff coordinate with the ARCS team for access and set up. Researchers may contact CQLS HDI staff to ask about the suitability of Sherlock Secure Cloud and for planning health data and informatics research.
Centralized Local REDCap Software for Data Collection Now Available
For researchers who may not require secure cloud computing, but plan to collect data may now access REDCap on a centralized local OSU server managed by CQLS. REDCap provides features not available in other data management systems including support for large-scale longitudinal studies. CQLS offers free OSU-specific training resources in addition to the REDCap Consortium resources for REDCap users. Use of REDCap locally for research projects is available on a fee-basis. Researchers should review materials on the CQLS website and contact CQLS to set up REDCap accounts for their research teams and to consult on their data management plans.
High-quality reference genomes are the foundation for much of what our collaborators do at OSU — from mapping reads and calling variants to identifying genes of interest and performing CRISPR knockouts. Obtaining high-quality genomes is still an evolving field: only recently did NIH researchers from the Telomere-to-Telomere consortium bring to full completion a human reference genome; this milestone led to the discovery of nearly 2,000 additional gene predictions, broadly improving our ability to understand genetic variation and epigenetic activity (Sergey Nurk et al. 2022). At the forefront of this achievement was developing protocols for Nanopore ultra-long (UL) sequencing: critical for spanning centromeres, telomeres, and other repeat-rich, heterozygous, and duplicated regions. At CQLS, we’re adapting this methodology for plants to overcome technical biases that persist in traditional short-read and long-read sequencing.
Traditional methods for plant DNA extraction typically rely on aggressive homogenization and lysis methods to break down cell walls, resulting in highly fragmented DNA. Bead beating, vortexing, and even pipette mixing can shear long DNA molecules. By isolating intact nuclei and removing cytoplasmic contaminants without rupturing the nuclear membrane, we can recover DNA fragments that span >60 kilobases to 1Mbp ideal for UL sequencing. We’re pleased to report that our yield of UL reads is on par with other leading institutions.
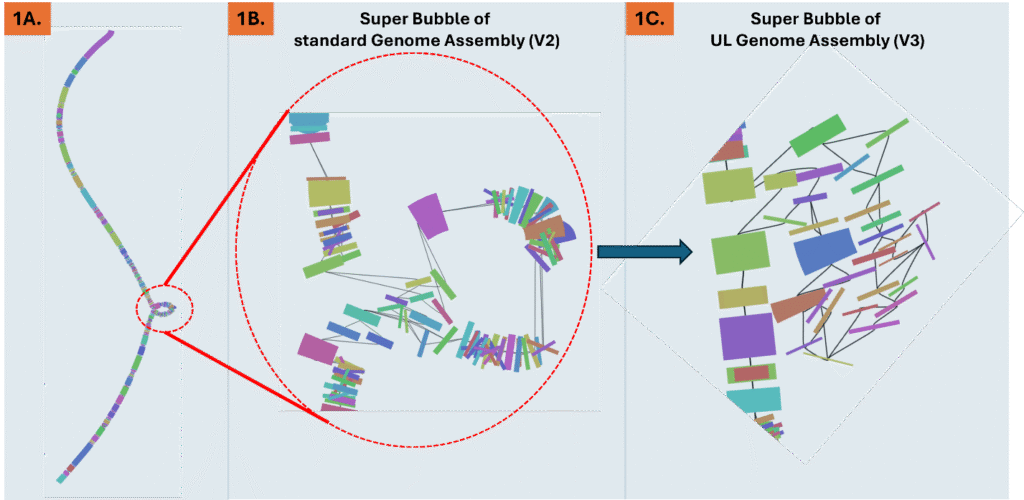
Figure 1. A. Visualizing of a DNA genome assembly graph (V2) that lacks ultralong reads. Different colors along the graph indicate Simple Bubbles that represent heterozygous loci. The dotted red circle is a Super Bubble composed of many bubbles at different positions within the allelic locus. B. Magnification of the dotted-red circle Super Bubble from a genome assembly that lacks the ultralong reads. C. The same Super Bubble from 1B. identified in the ultralong assembly (V3) shows a significant reduction in the number of bubbles within, vastly reducing complexity of heterozygous calls and read mapping at this allelic locus.
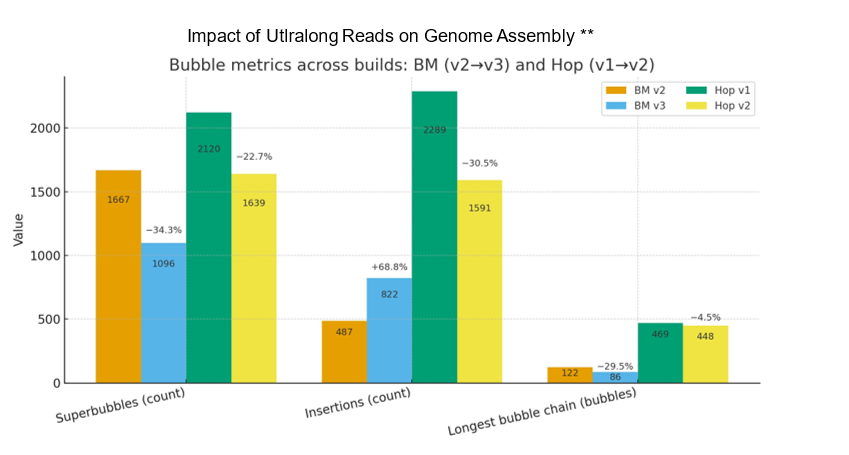
Even modest UL coverage simplifies assemblies and improves accuracy. In benchmarks on a polyploid mint and highly heterozygous hop, ~5x UL depth reduced the total number of spurious short contigs by three-fold compared to genomes without UL support. To illustrate this, Figure 2A. shows a genome assembly graph that lacks UL support. Complex regions in the graph often contain numerous heterozygous loci that exist within a larger heterozygous locus (known as a super bubble). Comparing the same super bubble between two genomes, one without ultralong (Figure 1B) and one with 5x UL coverage (Figure 1C), we can see a significant reduction in total bubbles and complexity. Across the entire genome, UL assemblies reduce super bubbles by ~30% versus no-UL assemblies (Figure 2). These structural improvements carry through to the chromosome validation, yielding cleaner Hi-C contact maps that contain less technical artifacts and stronger assembly metrics overall.
Figure 2. Impact of ultralong reads on raw genome assembly graphs of two plant species. A polyploid species of mint is represented in orange (raw assembly) and blue (UL reads). Highly heterozygous hop in green (raw assembly) and yellow (UL reads). Plots represent three distinct categories of assembly graph statistics: the number of super bubble, the number of insertions within bubbles and the total number of sequential bubbles.
Ultralong reads deliver biological insights by resolving previously unknown regions that can erroneously impact downstream analysis. Many facets of gene function and regulation have been found to be embedded within or adjacent to transposable-element rich regions that are difficult to assemble correctly without ultralong support. In mint, UL reads led to a nearly seven-fold reduction in an expanded higher-order nucleotide repeat, greatly clarifying our understanding of a centromeric region. We expect UL data to continue strengthening our understanding of biology, especially in non-model species that may hold unknown complexity.
References
Sergey Nurk et al. ,The complete sequence of a human genome.Science376,44-53(2022).DOI:10.1126/science.abj6987
Registration is free this year. Lunch is optional.
Friday November 22, 2024
PROGRAM
Time
Event
8:00 – 9:00 AM
Registration, poster & sponsor set-ups, coffee and light refreshments
9:05 – 9:15 AM
Brent Kronmiller, Interim Director, CQLS “CQLS Introduction and updates”
9:15 – 9:50 AM
“Lightning Round” Research Presentations
9:50 – 10:20 AM
Break – Poster setup and Sponsor displays
10:20 – 11:15 AM
Key Note Speaker, Moderator: Siva Kolluri Joe Gray, Biomedical Engineering, OHSU“Rethinking Precision Cancer Treatment for Advanced Cancers – A Systems Biomedicine Approach”
11:15 AM – 12:00 PM
OSU Faculty Research Presentations Moderator: Chrissa Kioussi
Olena Taratula, Pharmaceutical Sciences, OSU“Nanotheranostics for potential management of ectopic pregnancy”
Alex Weisberg, Botany and Plant Pathology, OSU“Evolution and epidemiology of global populations of Agrobacterium vitis”
12:00 – 1:00 PM
Lunch – Poster setup and Sponsor displays
1:00 – 2:30 PM
CQLS Core services Moderator: Brent Kronmiller
CQLS Bioinformatics Staff“CQLS Sequencing and Bioinformatics Consulting Services” Laurent Deluc, Horticulture, OSU“Sequencing of the Microvine Genome: an example for genome editing studies and modern genomic data resources”
Denise Hynes and Joe Spring, CQLS“CQLS Health Data Informatics Services” Veronica Irvin, School of Human Development and Family Sciences, OSU“Be Well: An example of using REDCap for NIH-funded studies”
Anne-Marie Girard, CQLS“CQLS Confocal Microscopy Services” Maria Camila Medina, Botany and Plant Pathology, OSU“From cell to shapes: Confocal insights into plant development”
2:30 – 3:00 PM
Break – Poster setup and Sponsor displays
3:00 – 4:10 PM
OSU Faculty Research Presentations Moderator: Fritz Gombart
Brandon Pearson, Environmental and Molecular Toxicology, OSU“Non-cancer health outcomes for environmental mutagens”
Maria Purice, Biochemistry and Biophysics, OSU“Impact of sex and aging on glia-neuron interactions in C. elegans”
Erin McParland, College of Earth, Ocean, and Atmospheric Sciences, OSU“Chasing the elusive chemical currencies of marine microbes with metabolomics”
4:10 – 4:20 PM
Irem Tumer, VPR, Division of Research and Innovation
The CQLS has experienced significant change in the last few years. Throughout this we have increased CQLS staffing, developed new research collaborations, and continued CQLS services without interruption. As we move forward the CQLS will be increasing community outreach and events including restarting the Fall Conference (November 22nd) and working with faculty committees and our users and community to direct the future of the CQLS.
The CQLS is composed of four interconnected functional groups:
CQLS Core Laboratory – Four laboratory staff run the services provided in Genomics Core, Shared Instrumentation Core, and Microscopy Core. Recently the laboratory has been working with the Division of Research and Innovation to move CQLS into the new RELMS instrument billing system.
CQLS Research Computing HPC – Two CQLS staff run and maintain the CQLS HPC, the largest and most widely used computing cluster at OSU. Recent upgrades outlined in this newsletter combine the CQLS HPC with other OSU HPC to create a consolidated OSU research computing cluster.
CQLS Bioinformatics and Data Science – Six CQLS staff consult on computational research and teach our computing and bioinformatics courses. CQLS scientists consult on experimental design, grant proposals, research, and programming projects and are highly utilized in many labs across OSU.
CQLS Health Data Informatics – Led by Denise Hynes, the CQLS provides support and tools for analyzing health data including protected and secure data.
In this newsletter you will find information on CQLS changes and new services, research projects in the CQLS, spotlight on CQLS service, and information on upcoming CQLS events.
After over a year of planning and hard work, CQLS is completing the upgrade of its entire high-performance computing (HPC) infrastructure to create a consolidated campus-wide HPC and provide accessible research computing across OSU.
The old GENOME cluster is being upgraded and replaced by an improved infrastructure with new features and across-the-board updates. These upgrades include new operating systems, Rocky 9 or Ubuntu 22, many software package updates, and improved resource management.
The Wildwood cluster uses SLURM as its primary job queueing system, which provides priority queuing and GPU-aware resource management. Wildwood also has SGE job management, to support those who rely on the SGE work flow for their analysis. To support multiple job management tools, we have also developed a suite of new tools (hpcman) that allow users to submit jobs and manage their work on SLURM or SGE with a unified command set.
One of the other big improvements in Wildwood is that we can now use ONID authentication – users no longer have to maintain a separate CQLS password, they can log in using their ONID password.
Wildwood is also a federated cluster – Wildwood connects to COEAS computing resources, and will soon also connect to the COE HPC. A single log in will allow users to run jobs and manage their data on any of these clusters, and shared storage provides a unified interface to research data.
The CQLS Wildwood HPC contains 9PB of data storage, ~6500 processing CPUs, and 80+ GPUs.
Labs and departments have been making the transition to Wildwood through the summer, but Wildwood also has an expanded ‘all.q’ – general access resources anyone can use, including GPUs and PowerPC architecture machine.
CQLS has installed a new Leica Stellaris 5 Confocal Microscope System which replaces an older confocal system. With this type of microscope one can obtain 3D sectioning of fluorescently labeled cells, or tissues for clearer, sharper images of specimens. People have used confocal systems to examine structures within living or fixed cells and to examine the dynamics of cellular processes.
3D rendering of veins in maize leaves. Yellow: Pin1a-YFP in cell membrane, Red: DR5-RFP in endoplasmic reticulum. Image courtesy of Camila Medina.
A confocal system has the capacity to image in Z and time to better visualize location in 3D than widefield fluorescence microscope by using a pinhole to eliminate out of focus light. The system has a white light laser (WLL) with tunable excitations from 485 nm up to 685 nm in addition to a 405 nm laser and sensitive HyD S detectors with a detection range from 410 to 850 nm. Additionally, the Stellaris system also has TauSense, a set of tools based on fluorescence lifetime information with potential to eliminate autofluorescence, and LIGHTNING which expands the extraction of image details for both classical imaging range and beyond the diffraction limit (120nm).
We will be offering free training and imaging time during this fiscal year to those people who have a project ready for imaging and in order to help with grant writing for future imaging projects. Contact Anne-Marie Girard to discuss potential projects or for more information about the system or its capacities.