It is my pleasure to announce that Dr. Kathryn A. Higley has been selected to serve as the Interim Director of the Center for Quantitative Life Sciences (CQLS) effective December 1.
Dr. Higley has been at OSU for 27 years, and most recently served as the Associate Director for the TRACE-OSU effort. She has a long record of both administrative and academic experience at OSU. She is a Professor and former Head of the School of Nuclear Science and Engineering in the College of Engineering at Oregon State University. She has managed OSU’s Radiation Health Physics program, including developing its online graduate degree into the largest in the country. Dr. Higley has been at Oregon State University since 1994 teaching undergraduate and graduate classes on radioecology, radiation biology, and more. She is a Council Member of the National Council on Radiation Protection and Measurements, a board member and fellow of the Health Physics Society, and a Certified Health Physicist. Dr. Higley and her students have done research in radiologically contaminated environments around the globe.
I am excited to start working with Dr. Higley, CQLS staff and affiliated faculty to take the center to the next stage. Please join me in welcoming her to the CQLS and RO community!
Sincerely, Irem Y. Tumer, Ph.D., ASME Fellow Vice President for Research
Another great term of the CGRB’s Bioinformatics User Group (BUG) is in the books!
This term we had a wide range of presenters—graduate students to Principle Investigators. It was nice to get the perspective of folks who are in different parts of their careers.
A special thanks to all of our presenters:
Sept 25: Christopher Sullivan and Ken Lett (Center for Genome Research & Biocomputing)
Title: CGRB’s new DFS for one and all!, i.e., Don’t know what a Distributed File System is? Come find out!
Abstract: The CGRB works with researchers to provide the most robust computational infrastructure available today. Many group rely on file services at the heart of their research computing needs and the CGRB has worked for over 2 decades to provide redundant high speed file services. Over the years users have grown to expect the best solution at a very cheap price. Because of this model the CGRB spends a great deal of time evaluating the available systems to ensure we always have the best at the lowest price. In the past year the CGRB has worked to evaluate and purchase new file service hardware that will replace our existing setups. We will be explaining the pathway taken to bring the new service online and some of the new exciting features.
Oct 9: Lillian Padgitt-Cobb (David Hendrix Lab, Biochemistry & Biophysics)
Title: A phased, diploid assembly of the hop (Humulus lupulus) genome reveals patterns of selection and haplotype variation, i.e., Resolving functional and evolutionary mysteries of a large, complex plant genome with genomic data science
Abstract: Hop (Humulus lupulus) is a plant valued for its use in brewing and traditional medicine. Efforts to determine how biosynthetic pathways in hop are regulated have been challenged by its complex genomic landscape. The diploid hop genome is large, repetitive, and heterozygous, which challenged early attempts at sequencing with short-reads. Advances in long-read sequencing have improved detection of repeats and heterozygous regions, revealing that the genome is nearly 78% repetitive. For our assembly, PacBio long-read sequences were assembled with FALCON and phased into haplotype assemblies with FALCON-Unzip. Using the phased, diploid assembly to assess haplotype variation, we discovered genes under positive selection enriched for stress-response, growth, and flowering functions. Comparative analysis of haplotypes provides insight into large-scale structural variation and the selective pressures that have driven hop evolution. The approaches we developed to analyze the phased, diploid assembly of hop have broader applicability to the study of other large, complex genomes.
Oct 23: Kelly Vining (Kelly Vining Lab, Horticulture)
Title: R/qtl, i.e., Applications and methods for analysis of quantitative traits
Abstract: R/qtl is an R package that is used for genetic mapping and marker-trait association. This presentation will explore specific features of R/qtl applied to plant breeding populations. Data types, functions, and interpretation of results will be explored.
Nov 6: Ed Davis (Center for Genome Research & Biocomputing)
Title: Introductory microbiome analysis using phyloseq, i.e., How to generate exploratory diversity plots and what they mean
Abstract: Generating high quality, publication ready figures for a microbiome study can be somewhat difficult. An understanding of both the statistical tests and how to effectively use R to produce figures is required, so the learning curve can be somewhat steep. Fortunately, there are several easy-to-use packages in R that facilitate the analysis of microbiome studies using 16S amplicon data, including the phyloseq package that will be the focus of my talk. I will cover the basics of analyzing alpha and beta diversity and provide some code and example images to show how to generate publication ready figures starting from the base phyloseq output. I will also generate some exploratory charts and graphs such that one would be able to form and later test hypotheses using microbiome data. I will be happy to share the examples and code as well, so that I might catalyze the analysis of your own microbiome studies.
Nov 20: Cedar Warman (John Fowler Lab, Botany & Plant Pathology)
Title: High-throughput maize ear phenotyping with a custom-built scanner and machine learning seed detection, i.e., Computer counts corn, correctly.
Abstract: Near-incomprehensible amounts of maize are produced each year, but our understanding of the dominant North American crop is fundamentally incomplete. Of particular interest is the seed-producing structure of maize, the ear. Here, we present a novel maize ear phenotyping system. Our system captures a video of a rotating ear, which is subsequently flattened into a projection of the ear’s surface. Seed positions and genetic markers can be quantified manually from this projection. To increase throughput, we applied deep learning-based computer vision approaches to seed and marker quantification. Our progress towards a completely automated phenotyping system will be described, in addition to challenges we continue to face adapting computer vision technology to maize ears.
Dec 4: Christina Mulch (Kelly Vining Lab, Horticulture)
Title: IsoSeq pooling and HiSeq multiplexing comparison for Rubus occidentalis samples to explore Aphid resistance, i.e., Utilizing RNA to find differences between Aphid Resistant and Susceptible plants.
Abstract: Black raspberry (Rubus occidentalis L.) is a small specialty crop produced primarily in the Pacific Northwest of the U.S. A major challenge for its success is Black raspberry necrosis virus vectored by the Large Raspberry Aphid (Amphorophora agathonica A.). We used Pacific Biosciences IsoSeq long read sequencing technology to study the gene expression patterns in leaves following aphid inoculation. We collected samples from a segregating population for resistance to the pest. High quality RNA was extracted from 20 samples, 10 resistant (R) and 10 susceptible (S) using a modified RNA extraction protocol. Data processing was preformed using the IsoSeq3 pipeline. Alignment of each R and S pool to the latest chromosome level black raspberry reference genome used minimap2 according to recommended options for IsoSeq. Reads were filtered based on mapping quality, alignment length, and presence or absence in multiple samples. This study seeks to reveal the genetic underpinning of aphid resistance with the ultimate goal of enabling marker assisted selection.
Thank you for attending and we look forward to seeing you in
2020!
All of the slots for winter 2020 are full, but please contact us if you’re interested in presenting in the future.
Aaron Trippe discusses the changes and challenges of working with the PacBio Sequel since 2016. He discusses improvements in the technology since 2016 and has advice for user who would like to utilize this service.
Aaron Trippe, our long-time PacBio technician, stands next to the CGRB’s Pacific Biosciences Sequel.
Q1: How long have you been running the PacBio sequencing service at the CGRB?
The CGRB was one of the early adopters of the Sequel, the second phase of long read genomic sequencing technology from Pacific Biosciences. It arrived here on campus in August of 2016. Since then the technology has made significant improvements to the user-interface, and has tremendously increased read lengths and output.
Q2: You started up the PacBio sequencing service at the CGRB. What has been the most challenging aspect about developing this service?
Aside from the continually changing and evolving technology, one of the most challenging aspects of the service is getting everything you feed the machine to produce optimal results. One of the advantages of the technology is that you are sequencing native DNA, but that also makes it challenging when working with an organism that traditionally is difficult to work with and considered problematic. Finding ways to produce super clean and high molecular weight DNA from just about everything is probably the largest hurdle to working with the technology as a service provider. The keys to success are definitely within the sample quality. Having pure, high molecular weight DNA is essential to take advantage of the long read aspect of the technology, and is directly correlated to the quality of the sequencing output.
Q3: What type(s) of project(s) would you recommend to use PacBio’s long read technology?
The technology is great for just about any sequencing application. With the long reads, you have access to regions of DNA that were not previously accessible due to repetitive regions in genomic DNA. There is enough output to multiplex several microbial genomes on a single SMRT Cell. Complete sequences of multiplexed amplicons using Circular Consensus Sequencing for high fidelity reads of shorter inserts. With the read lengths exceeding that of RNA transcripts, Isoform sequencing using the Iso-seq application is also available for obtaining complete transcripts.
Q4: Favorite or most interesting project you’ve worked on?
Since managing the PacBio Sequel, I’ve gotten to work with plants, animals (vertebrates/invertebrates), fungi, bacteria, and insects for the local scientific community, and beyond. I can’t say that I have had a favorite organism, and they have all been interesting projects, but overcoming challenges with successful results always feels rewarding.
Note: We wish Aaron the best as he purses a new opportunity and are grateful he was able to develop a successful PacBio Service at the CGRB! For future sequencing inquires please contact Katie Carter.
The CGRB will be offering three different workshops this fall. For more information and to register, see the CGRB website.
All workshops are available for credit for students or available to non-students as non-credit workshop(s).
To give perspective students a better insight on each course, we’ve conducted short interviews with the instructors about their course.
See course descriptions and the interviews with the instructors below!
Courses Offered:
Introduction to Unix/Linux and Command-Line Data Analysis (2 modules x 5 weeks @ 2 hrs per week)
Instructor: Matthew Peterson
Course Description:
Introduction to Unix/Linux (5 weeks @ 2 hrs per week)
Logistics: Date & Time: Sep 25 – Oct 23, Mon/Wed 2:00pm – 2:50pm For credit: BDS599 CRN 20579 Workshop Cost: $250 This module introduces the natural environment of bioinformatics: the Linux command line. Material will cover logging into remote machines, filesystem organization and file manipulation, and installing and using software (including examples such as HMMER, BLAST, and MUSCLE). Finally, we introduce the CGRB research infrastructure (including submitting batch jobs) and concepts for data analysis on the command line with tools such as grep and wc.
Command-Line Data Analysis (5 weeks @ 2 hrs per week)
Logistics: Date & Time: Nov 4 – Dec 4, Mon/Wed, 2:00pm – 2:50pm For credit: BDS599 CRN 20580 Workshop Cost: $250
The Linux command-line environment has long been used for analyzing text-based and scientific data, and there are a large number of tools pre-installed for data analysis. These can be chained together to form powerful pipelines. Material will cover these and related tools (including grep, sort, awk, sed, etc.) driven by examples of biological data in a problem-solving context that introduces programmatic thinking. This module also covers regular expressions, a useful syntax for matching and substituting string and sequence data.
Matthew Peterson in the CGRB server room
Q1: What do you hope students gain from this workshop?
My hope is that students come to appreciate the power and flexibility of using the text-based command-line interface to interact with (Linux) computational infrastructures. With practice students will become self-sufficient in utilizing the infrastructure to conduct their own research.
Q2: Favorite topic in your course?
Pipelines! The ability to chain the inputs and outputs of multiple commands to filter data is immensely powerful.
Q3: Who should register for this course?
From the first page of the course syllabus: “Linux/Unix Commands, Bioinformatics Utilities, Computational Infrastructure: If you know nothing about the above, then you are exactly in the right course! WELCOME!”
Q4: Advice for users new to bioinformatics and/or programming?
Practice, practice, practice! Learning how to use the command-line effectively is like making a clay pot, you need to get your hands dirty!
RNA-Sequencing (10 weeks @ 2 hrs per week)
Instructor: Dr. Andrew Black
Course Description:
Logistics: Date & Time: Sept 25 – Dec 5, Tue/Thur 11:00am – 11:50am For credit: BDS 599, CRN 20581 Workshop Cost: $500
This course provides an introduction to, and practical experience with, the computational component of bulk-RNA-sequencing. After a general overview, participants will obtain a working introduction to command line, R-studio, and accessing and utilizing a computing infrastructure. Students with then work through a series of exercises cleaning raw FASTQ files, aligning reads to a reference genome, quasi-mapping reads to a transcriptome / de novo assembly, followed by data visualization and Differential Gene Expression analysis.
Dr. Andrew Black will teach the RNA-seq workshop this term.
Q1: What do you hope students gain from this workshop?
I hope that students gain an understanding of the computational workflow involved with RNA-seq and an appreciation of the methodology! My overarching goal with this course is that people can use material from this course as scaffolding for analyzing their own data on the CGRB infrastructure.
Q2: Favorite topic in your course?
I added a lord of the rings theme to my course; students are looking for differentially expressed genes between hobbits and golems. I’m a dork, I know, but I had fun spiking different genes into the data and enjoy having students visualize this.
Q3: Who should register for this course?
Graduate students, postdocs, faculty, or anyone outside of OSU that are interested in receiving an introduction to RNA-seq or for those that are needing to learn the workflow for their own project(s).
Q4: Advice for users new to bioinformatics and/or programming?
Take it one step at a time and get comfortable with several commands before expanding your scope. Also, record your commands / code in a text document, because if you aren’t using it on a daily basis, you’ll forget it!
Data Programming in R (6 weeks @ 3 hrs per week)
Instructor: Dr. Shawn O’Neil
Logistics: Date & Time: Sept. 25 – Nov. 6, Mon/Weds/Fri 9:00am – 9:50am For credit: ST 599, CRN 17196 Workshop Cost: $500
The R programming language is widely used for the analysis of statistical data sets. This course introduces the language from a computer science perspective, covering topics such as basic data types (e.g. integers, numerics, characters, vectors, lists, matrices, and data frames), importing and manipulating data (in particular, vector and data-frame indexing), control flow (loops, conditionals, and functions), and good practices for producing readable, reusable, and efficient R code. We’ll also explore functional programming concepts and the powerful data manipulation and visualization packages dplyr and tidyr, and ggplot2.
Q1: What do you hope students gain from this workshop?
I really hope that students gain an appreciation for programming as a creative activity. It’s not just a means to an end, even with a statistical language like R; there’s a lot of room for play and exploration. Simulation, for example, is a great way to explore complex systems and ask ‘what if’ questions. Many languages (including R) support programmatic drawing and data visualization which can be quite fun.
Q2: Favorite topic in your course?
I always enjoy the point when we first start scaling analyses to thousands of statistical tests. It’s an eye-opening moment, and doing so in R introduces ‘functional programming,’ a powerful and increasingly important paradigm for software design.
Q3: Who should register for this course?
Anyone who is interested in doing data analysis, especially of a statistical sort. For those interested in learning programming in a broader sense, our winter Intro to Python series is an excellent overview of fundamental concepts. Although we cover the same topics in the R course, R organizes its features differently than most mainstream programming languages like Python, Java, and C++. Learning both Python and R provides a solid foundation for data science!
Q4: Advice for users new to bioinformatics and/or programming?
I do recommend learning more than one programming language, eventually, as this helps separate deeper concepts from syntax. Find what motivates you and explore it via programming — this could be your primary research project, some field you’ve been wanting to learn more about, or even a hobby.
The CGRB’s biocomputing infrastructure was highlighted in AMD CEO Lisa Su’s keynote speech at the Consumer and Electronics Show (CES) in Las Vegas on January 9, 2019. Watch below:
This blog post was originally published on September 10, 2018 and written by Christopher M. Sullivan, Assistant Director for Biocomputing. Read the whole article here.
The Oregon State University’s Center for Genome Research and Biocomputing (CGRB) and the Plankton Ecology Lab at OSU Hatfield have been collaborating in implementing an image processing pipeline to automate the classification of in situ images of plankton: microscopic organisms at the base of the food web in the world’s oceans and freshwater ecosystems. The imagery collection from a 10-day cruise typically contains approximately 80 TB worth of video, which, in some cases, may convert into image data yielding several billions of segments representing individual plankton and particles that need to be identified; a near impossible task to carry out manually by human experts. While we have a fully functional Convolutional Neural Net (CNN) algorithm that does an excellent job at predicting the identity of the plankton organisms or particles, we have been limited by GPU computational capabilities. We started working with PCI bus based Tesla K40 and K80 GPUs, which were good enough to manage millions of segments. However, when it came to billions of segments, it became a near insurmountable challenge.
R’s default print function for data frames and matrices is not an effective way to display the contents, especially in a html report. RStudio created a R package, DT, that is an interface to the JavaScript library DataTable. DT allows users to have interactive tables that includes searching, sorting, filtering and exporting! I routinely use these tables in my analysis reports.
Install the DT package from cran
First, one must install and load the DT package. Open up RStudio and run the following commands to install and load the DT package:
# Install the DT package
install.packages("DT")
# Load the DT package
library(DT)
Example Table
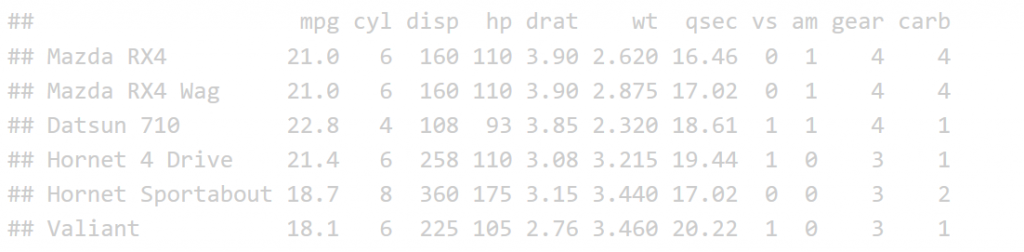
The print function is not the most effective was to display a table in an HTML R Markdown report.
print(head(mtcars))
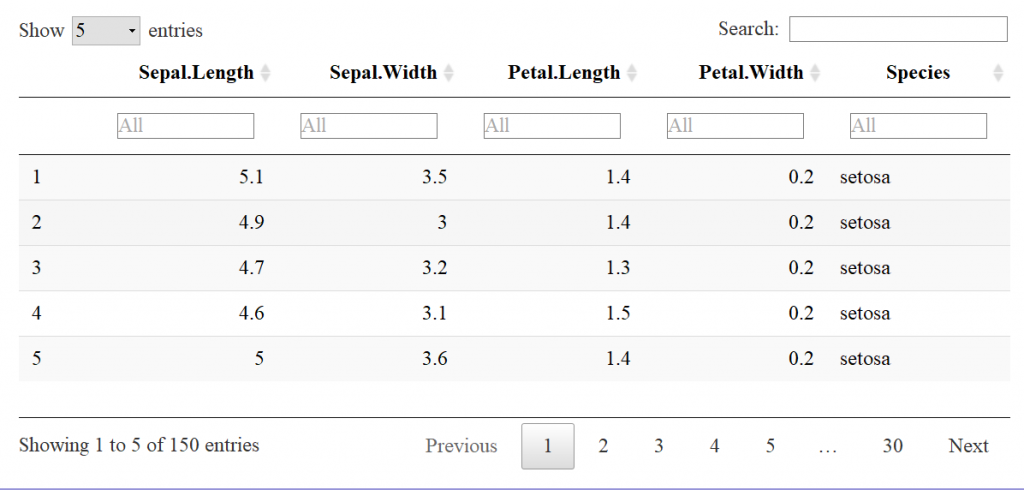
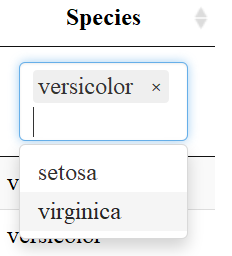
Now let’s look at the datatable function for comparison. The input to the datatable function is a data frame or matrix. Let’s make a table with the preloaded iris data that’s in a data.frame. The basic call is DT::datatable(iris) but in our example I’ve added the filter option to the top of the table, and limited the number of entries to 5 per table. See code and table features below:
Already, the readability is much better than the base r function print. This is a JavaScript based table, stored in a HTML widget, so a flat image doesn’t convey all of the interactive features.
Features
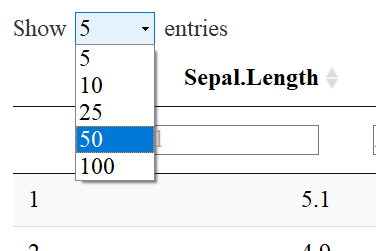
NUMBER OF ENTRIES TO DISPLAY
You’ll notice that there is a drop down menu that says: “Show 5 entries”. The default is 10, but I specified 5 as default with the code pageLength=5. One may select the number of entries to show by using the drop down menu like so:
SEARCH BAR
The widget also includes a search bar on the top right corner which can be very useful when interactively exploring data. Note at the bottom of the table it shows you how many entries (rows) were found and are being displayed.
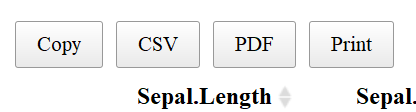
SORT COLUMNS
Notice that to the right of each column name are two arrows: One may sort by ascending or descending order and the direction of the blue arrow indicates by which direction you sorted the column.
FILTER COLUMNS
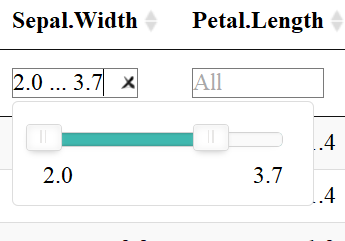
The datatable function also allows users to filter each column depending on the datatype: filter numeric columns with a slider & filter columns of class factor with a drop down menu. One must add the filter = "top" (or bottom, etc.) to the code to enable this feature.
Numeric columns have slidersColumns of class factor have a drop down menu
Export Data
Another useful aspect of the datatable function is the “Buttons” extension. This enables users to copy the table, save as a csv, excel or PDF file, or print the table. The table “remembers” what you’ve changed so far—so if you sort by Sepal Length, filter pedal width to > 1 and select species “versicolor” the copied/saved table will have these same restrictions.
The above code adds “buttons” to the top of the table like so:
If one clicks “copy”, the table will be copied to your clipboard, “CSV” or “PDF” will save the table to the give file type, and “print” will bring put the table into a print friendly format and will bring up the print dialog box.
Links and Color
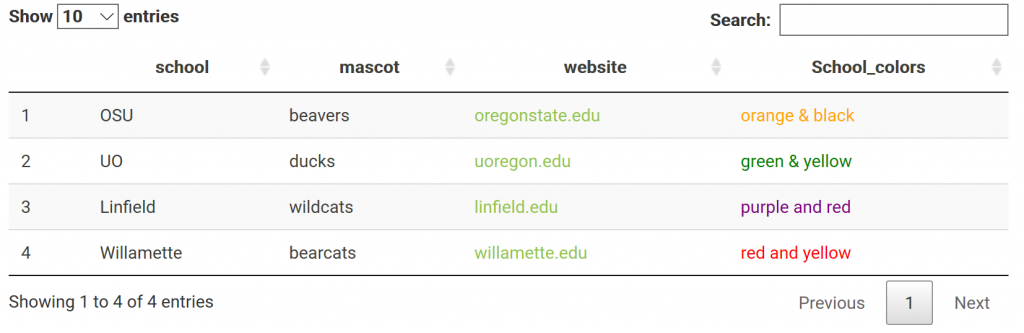
One may also have links in their table. Say you made a data frame with links you want to work in your html report. For example: a data frame of variants w/ links to their position in a genome browser. This is done through not escaping content in the table, specifically the column with the links. The links are made with html and must not be escaped to show up. This applies to other html as well; including color. For me, it was confusing that I had to not escape the html columns. Got it completely backwards the first time I tried it. NOTE: > got replaced with “& gt;” (with no spaces) when it is rendered on the blog… Need to find a fix!
# Make dataframe
df.link <- data.frame(school=c("OSU", "UO", "Linfield", "Willamette"),
mascot=c("beavers", "ducks", "wildcats", "bearcats"),
website=c('<a href="http://oregonstate.edu/">oregonstate.edu</a>',
'<a href="https://www.uoregon.edu/">uoregon.edu</a>',
'<a href="https://www.linfield.edu/">linfield.edu</a>',
'<a href="https://www.willamette.edu/">willamette.edu</a>'),
School_colors=c('<span style="color:orange">orange & black</span>',
'<span style="color:green">green & yellow</span>',
'<span style="color:purple">purple and red</span>',
'<span style="color:red">red and yellow</span>'))
# When the html columns, 3 & 4, are not escaped, it works!
datatable(df.link, escape = c(1,2,3))
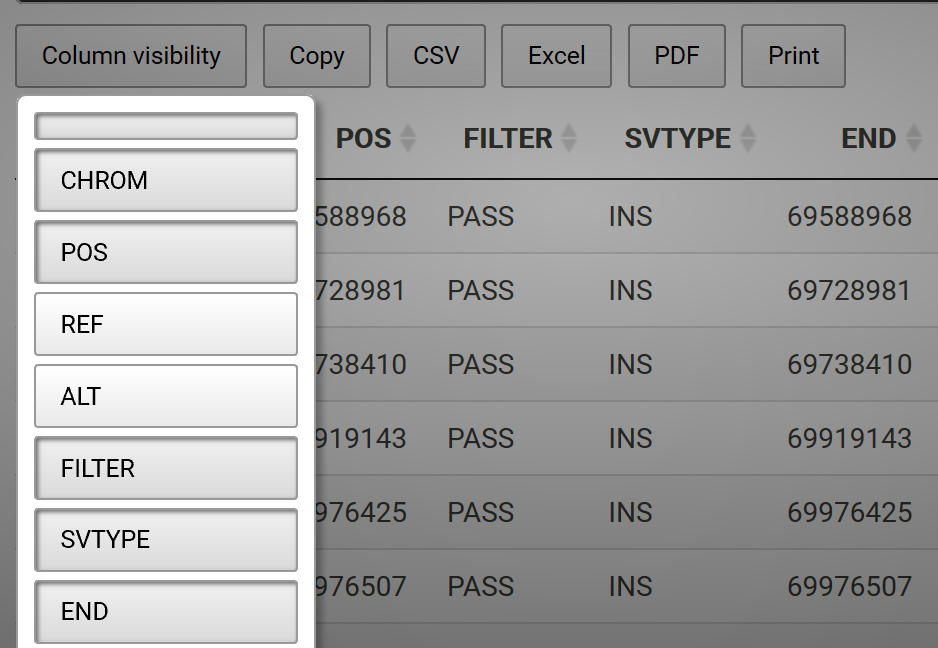
Column Visibility
One may also hide columns from visibility and add a button to add the column back interactively. For example, say we have a data frame called sv.all.i.in. We can hide columns 3 and 4, which are long sequences and disrupt the readability of the table, with the following code:
The CGRB 2019 Fall conference registration is now open! Please join us for our annual event this September for informative talks, posters and a reception. This year the Fall Conference will also include lighting talks.
Important Information:
When: Friday, September 20, 2019
Where: CH2M Hill Alumni Center – Oregon State University
Andrew Annalora, Environmental and Molecular Toxicology Exploring Splice Variant Biology in Nuclear Receptor and Cytochrome P450 Genes
9:40 – 10:30
Ed Kelly, University of Washington Organs on a Chip – Chips in Space
10:30 – 10:55
Break (Poster and Sponsor displays)
10:55 – 11:35
Morning Lightning Talks (8 talks) – moderated by Jeff Anderson
11:35– 12:00
Felipe Barreto, Integrative Biology Genomics in the Tidepool: Functional and Population Genetics of Adaptation and Speciation in a Tiny Crustacean
12:00 – 12:25
Kevin Brown, College of Pharmacy Adventures in Complex Systems
12:25 – 1:25
Lunch (Poster and Sponsor displays)
Afternoon Session
Hosted By Craig Marcus
1:25 – 1:50
Afua Nyarko, Biochemistry and Biophysics Selectivity and Specificity in Cancer Regulatory Proteins
1:50 – 2:40
Daniel Liefwalker, Oregon Health and Science University Therapeutic strategies targeting c-MYC
2:40 – 3:20
Afternoon Lightning Talks (8 talks) – Moderated By Viviana Perez
3:20 – 3:45
Break 25 mins (Poster and Sponsor displays)
3:45 – 4:10
Morgan Giers, Chemical, Biological, and Environmental Engineering Regenerating the Intervertebral Disc: Developing Effective Therapies in a Nutrient Limited Environment
4:10 – 5:00
Doris Taylor, Texas Heart Institute Building Solutions for Heart Disease: A 2019 Update
5:00 – 7:30
Poster Session / Reception, Sponsor Displays
Call for posters!
Invitation to present a Poster at the 2019 CGRB FALL CONFERENCE (Sept 20, 2019) Students, Post Docs, Research Staff and Research Faculty are invited to present their research as a Poster. Presenters are strongly encouraged but not required to consider utilizing a revolutionary new trend in poster format: https://twitter.com/mikemorrison
https://osf.io/ef53g (Posters in any format displayed at recent meetings are also welcome) Prizes for Best Posters: $100 (Undergraduate, Graduate and Post Doc Categories).
All fields and research topics welcome. To submit a Poster, please navigate to https://beav.es/ZPd DEADLINE Sept. 7, 2019.
Call for lightning talks!
Invitation to present a Lightning Talk at the 2019 CGRB FALL CONFERENCE (Sept. 20, 2019) https://cgrb.oregonstate.edu/fall-conference Students, Post Docs, Research Staff (FRA, Res. Associates, etc.), and Research Faculty are invited to present their research as a 5-minute Lightning Talk at the annual CGRB Fall Conference, Friday Sept. 20, 2019. First Prize for Best Lightning Talk = $100 Conference Registration Fee is waivedfor all Lightning Talk Presenters
All fields and research topics welcome. To submit a lightning talk: Please navigate to beav.es/ZPA
Talks are limited to 5 minutes and 5 slides maximum. Please Submit no Later Than: August 15, 2019. Talks will be selected by the Program Committee and Presenters notified by Aug. 31, 2019.
Committee Members
Thank you to our 2019 Fall Conference Committee:
Jaga Giebultowicz, Department of Integrative Biology Craig Marcus, Environmental and Molecular Toxicology Jeff Anderson, Department of Botany and Plant Pathology Viviana Perez, Department of Biochemistry and Biophysics