Sam Talbot, CQLS
High-quality reference genomes are the foundation for much of what our collaborators do at OSU — from mapping reads and calling variants to identifying genes of interest and performing CRISPR knockouts. Obtaining high-quality genomes is still an evolving field: only recently did NIH researchers from the Telomere-to-Telomere consortium bring to full completion a human reference genome; this milestone led to the discovery of nearly 2,000 additional gene predictions, broadly improving our ability to understand genetic variation and epigenetic activity (Sergey Nurk et al. 2022). At the forefront of this achievement was developing protocols for Nanopore ultra-long (UL) sequencing: critical for spanning centromeres, telomeres, and other repeat-rich, heterozygous, and duplicated regions. At CQLS, we’re adapting this methodology for plants to overcome technical biases that persist in traditional short-read and long-read sequencing.
Traditional methods for plant DNA extraction typically rely on aggressive homogenization and lysis methods to break down cell walls, resulting in highly fragmented DNA. Bead beating, vortexing, and even pipette mixing can shear long DNA molecules. By isolating intact nuclei and removing cytoplasmic contaminants without rupturing the nuclear membrane, we can recover DNA fragments that span >60 kilobases to 1Mbp ideal for UL sequencing. We’re pleased to report that our yield of UL reads is on par with other leading institutions.
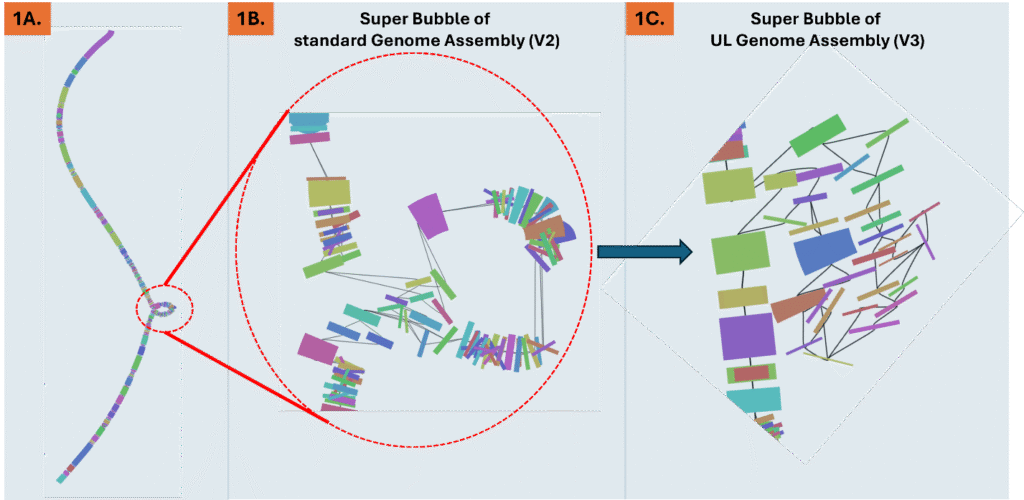
Figure 1. A. Visualizing of a DNA genome assembly graph (V2) that lacks ultralong reads. Different colors along the graph indicate Simple Bubbles that represent heterozygous loci. The dotted red circle is a Super Bubble composed of many bubbles at different positions within the allelic locus. B. Magnification of the dotted-red circle Super Bubble from a genome assembly that lacks the ultralong reads. C. The same Super Bubble from 1B. identified in the ultralong assembly (V3) shows a significant reduction in the number of bubbles within, vastly reducing complexity of heterozygous calls and read mapping at this allelic locus.
Even modest UL coverage simplifies assemblies and improves accuracy. In benchmarks on a polyploid mint and highly heterozygous hop, ~5x UL depth reduced the total number of spurious short contigs by three-fold compared to genomes without UL support. To illustrate this, Figure 2A. shows a genome assembly graph that lacks UL support. Complex regions in the graph often contain numerous heterozygous loci that exist within a larger heterozygous locus (known as a super bubble). Comparing the same super bubble between two genomes, one without ultralong (Figure 1B) and one with 5x UL coverage (Figure 1C), we can see a significant reduction in total bubbles and complexity. Across the entire genome, UL assemblies reduce super bubbles by ~30% versus no-UL assemblies (Figure 2). These structural improvements carry through to the chromosome validation, yielding cleaner Hi-C contact maps that contain less technical artifacts and stronger assembly metrics overall.
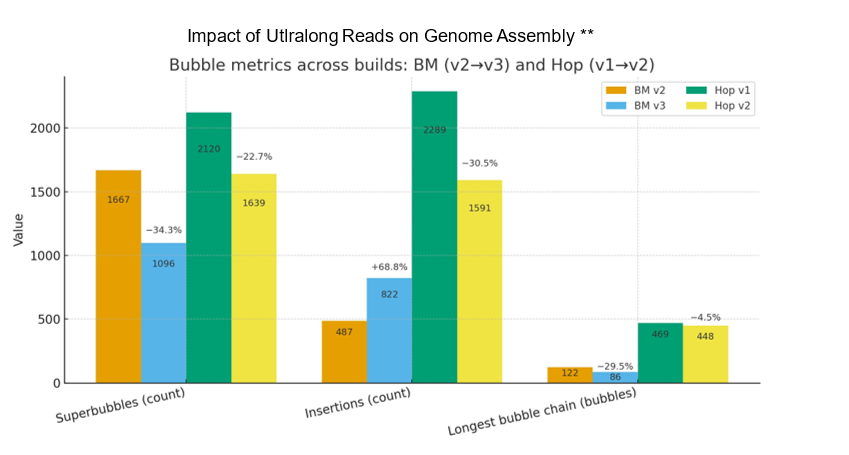
Figure 2. Impact of ultralong reads on raw genome assembly graphs of two plant species.
A polyploid species of mint is represented in orange (raw assembly) and blue (UL reads). Highly heterozygous hop in green (raw assembly) and yellow (UL reads). Plots represent three distinct categories of assembly graph statistics: the number of super bubble, the number of insertions within bubbles and the total number of sequential bubbles.
Ultralong reads deliver biological insights by resolving previously unknown regions that can erroneously impact downstream analysis. Many facets of gene function and regulation have been found to be embedded within or adjacent to transposable-element rich regions that are difficult to assemble correctly without ultralong support. In mint, UL reads led to a nearly seven-fold reduction in an expanded higher-order nucleotide repeat, greatly clarifying our understanding of a centromeric region. We expect UL data to continue strengthening our understanding of biology, especially in non-model species that may hold unknown complexity.
References
Sergey Nurk et al. ,The complete sequence of a human genome.Science376,44-53(2022).DOI:10.1126/science.abj6987